
R-2023-131
1.はじめに
新興科学技術をめぐっては、行政機関よりも実際に研究開発を担う民間企業や業界団体の側に多くの情報が蓄積されており、そこには明確な非対称性が存在する。そのため、適切な規制のフレームワークを設計するうえでは、被規制主体である民間からの情報提供が極めて重要である。規制主体と被規制主体のバランスを担保しつつ、被規制主体による適切な情報の提供を促すためには、両者の間における透明性をもったコミュニケーションと連携が実施されること、すなわちレスポンシブル・ロビイングの考え方を確立することが重要となる。
本稿では、医療分野、特に再生医療等製品を事例に規制の形成過程の変容と「レスポンシブル・ロビイング」の必要性について論じる[1]。
2.規制のラグと官民関係の変容
過去のReview[2][3]で論じたように、医療分野における革新的な製品・サービスをめぐっては、「規制のラグ」(Regulatory Lag)と呼ぶべき現象を見て取ることができる。ラグの発現の仕方は多様だが、共通しているのは規制主体の側が革新的な技術や価値を持った製品・サービスの登場に対して、それにタイムリーに対応できていないという現状である。実際、規制主体の側が先見的に既存の規制の変更や新たな規制の導入の必要性を認識し、適切なタイミングで規制を実行できるように備えることは、極めて高度に技術的な試みである。実際に具体的な規制対象となる製品・サービスが登場し、既存の規制との不整合が具体的に認識された段階になってようやく評価方法を含めた対応策を検討するようでは、文字通り「遅きに失する」ことになってしまう。そのため、技術開発の動向やコマーシャライズのタイミングを見据えつつ、技術的な成熟度合いに合わせてタイムリーに当該規制に関するルールの組成や変更を行わなければならない。それには、新たに登場することが見込まれる製品・サービスの特性が十分に明らかではないなかで、既存の規制との間でいかなる不整合が生じうるのか具体的に整理し、ルールづくりを進める必要がある。
こうした状況は、規制主体と被規制主体の関係性にも変化をもたらしつつある。従来は、規制主体の側がその時々の状況において求められる規制の内容・適用方法等を検討し、それを実際に適用する、いわばパターナリスティックな規制の立案と実施がなされてきた。しかしながら、今日の革新的な製品に対する規制の形成過程においては、規制主体が単独で規制内容を検討するのではなく、むしろ被規制主体の重大なコミットメントのなかで規制政策が形成される協調的な政策形成への移行が観察されつつある。
こうした変化の背景には、規制主体と被規制主体との間に新興科学技術を巡る情報の非対称性が深まっていることが挙げられる。規制主体が新興科学技術の特性について的確にモニタリングし、適時に的確な規制の導入や変更を講じることは現実的には困難である。そのため、実際の規制はイノベーターとの相互作用のなかで形成されており[4]、被規制主体は規制主体による規制の検討過程における機能不全を補完する存在といえる。被規制主体による技術特性に関する情報提供や当該技術に対する評価方法の提案こそが、新興科学技術に対する規制の質を左右する重要な要素となる。
3. 再生医療と「規制のラグ」
医療分野は、伝統的な規制政策の領域でありながら、技術革新に伴い被規制主体による規制主体に対する情報優位が進んだことを背景として、近年では規制主体と被規制主体との協調的な規制の検討と導入がみられる。以下では、協調的な規制の形成の事例として再生医療等製品の開発と規制の導入過程を確認する。
テルモ株式会社の再生医療等製品である骨格筋芽細胞「ハートシート®」は、日本発・世界初の心不全治療向け再生医療等製品であった。それゆえに、開発時点において審査側がその革新性に対する適切な評価手段を持ち合わせていなかった。そのため、審査当局である医薬品医療機器総合機構(PMDA)との事前面談、事前相談、申請前相談といった事前調整のプロセスから、実際に確認申請や治験届、承認申請といったフォーマルなプロセスに移行するのに多くの時間を要したことが知られている。
2007(平成19)年に大阪大学においてFirst in Human試験が実施され、テルモが細胞シートの開発に着手してから、2012(平成24)年に治験届が提出されるまでに約4年半、2014(平成26)年の承認申請までに約7年半、そして最終的に2015(平成27)年に薬事承認を取得するまでの間に実に約8年余りの時間を要することになった(表1および図1)。承認までのプロセスにおいてこれほど多くの時間を要することになったのは、ハートシートそのものの研究開発が遅れたのではなく、実際の技術的な成熟度合いの進展に比べて、規制主体の側における評価方法の構築が追いつかなったことが大きな要因として挙げられる。そのため、規制当局の側との事前調整がスムーズに進まず、治験の実施や承認申請までに膨大な時間を要することになった、まさに「規制のラグ」の代表的な事例といえる。
表1 ハートシートの開発および薬事承認までの経緯[5][6]
| 年月 |
内容 |
| 2000年 |
大阪大学が心筋細胞シート・筋芽細胞シートに関する基礎研究を開始 |
| 2003年 | 大阪大学とテルモが筋芽細胞の共同研究を開始 |
| 2007年5月 |
大阪大学がFirst in Humanとして補助人工心臓装着下の心臓移植待機患者に筋芽細胞シートを移植 |
| 2007年 |
テルモが細胞シートの開発に着手 |
| 2009年11月 |
PMDAとの事前面談を実施 |
| 2010年1月 |
PMDAに確認申請 |
| 2010年12月 |
PMDAとの事前相談を実施 |
| 2012年1月 |
テルモが技術移転を受け、PMDAに治験届を提出 |
| 2013年4月 |
再生医療推進法成立(議員立法) |
| 2013年11月 |
PMDAとの申請前相談を実施 |
| 2013年11月 |
再生医療等安全確保法および薬事法一部改正公布(2014年11月25日施行) |
| 2014年 |
治験の完了(国内3医療機関7例) |
| 2014年10月 | PMDAに承認申請 |
| 2015年9月 |
厚生労働省より製造販売承認を取得(条件及び期限付承認) |
出所:大阪大学およびテルモ(株)の各種発表資料より著者作成
図1 ハートシートの開発および薬事承認までの経緯
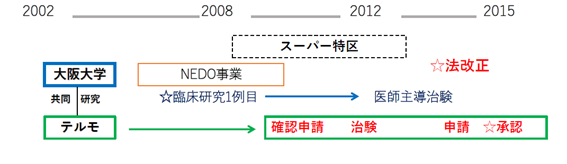
出所:大阪大学およびテルモ(株)の各種発表資料より著者作成
4. 官民協調的なルール形成
では、ハートシートをめぐる「規制のラグ」はどのようにして克服されたのだろうか。大きな転機となったのは、2013(平成25)年に施行された再生医療等安全確保法および薬事法の一部改正(いずれも11月27日に公布、翌2014年11月25日)である。再生医療等製品は、従来の医薬品とも医療機器とも異なる特性を持った極めて新しい技術領域であった。そのため、再生・細胞医療に関する研究開発の進展とともに、その製品化が目指されるようになるなかで、再生医療に固有の特性に応じた新たな規制のあり方が模索されることとなった。これらの法整備は、まさに再生医療の特性に応じた新たな規制の導入を目指したものであった。
特に注目するべきポイントは、こうした新たな規制のフレームワークを検討するうえで、研究開発の推進主体であるアカデミアと業界団体が特にアジェンダセッティングにおいて重要な役割を果たしたことである。具体的には、アカデミアである一般社団法人日本再生医療学会と2011(平成23)年6月に立ち上げられた業界団体である一般社団法人再生医療イノベーションフォーラム(FIRM: Forum for Innovative Regenerative Medicine)が規制主体の側に再生医療に関する知見が十分に蓄積されていないなかにあって、実用化と安全性確保の両面において議論をリードした。実際に、再生医療関連の法制化が進む駆動力となった再生医療推進法(2013年4月26日成立、議員立法)を端緒として、再生医療等安全性確保法および薬事法の一部改正のいずれについてもこれらの団体による積極的な関与がみられた[7]。
特に、再生医療という新しい技術のコマーシャライズにあたっては、製品・サービスとしての安全性の確保が大きな課題となっていた。当時は、自由診療における再生医療に対する規制が存在しておらず、2010年9月に京都Bethesdaクリニックにおける死亡事故が発生するなど、医師法上の医師の裁量に基づく再生医療の提供(幹細胞の輸注、投与、移植等)の品質、安全性等が社会問題化していた[8]。それを受け、日本再生医療学会が2011年1月に声明を出し、「患者の安全性を無視し日本医療の信頼を根幹から揺るがすような所謂、『未承認の再生・細胞医療』に対して医療法、薬事法等の改正等を推進し適切な新しい医療提供体制の構築による患者(国民)の安全性を早急に確保すること」を求めるなど、学術研究の推進という学会の役割を超え、政策提言にまで踏み込んでいる[9]。こうした取り組みをひとつの契機として、自由診療を含む再生医療に対する安全性の確保を目指した規制が具体的に検討されるようになった。
また、2014年の薬事法の一部改正(薬機法への変更)においては、「再生医療等製品」が新たな製品群として明確に定義されるとともに、その特性を踏まえた安全対策の規制が設けられることとなった。さらには、この改正においては、従来は事前の申請段階において確認を求められてきた安全性の確認を市販後に回すことで早期の実用化を可能とする「条件・期限付承認」(Conditional Early Approval System)という新たな仕組みが導入された。こうした早期の製品化や上市については、基礎研究や臨床を担うアカデミアのみならず、実際の製品開発を行う企業、そしてその業界団体が非常に強い問題・関心を持っていた。再生医療のような新たな技術群に対して、その有効性が確実に示されるまで承認ができないとなれば、先の「ハートシート」の事例にみられるように、コマーシャライズが大幅に遅れてしまうことになる。そうした事態を避けるための新たなアプローチとして、「条件・期限付承認」の可能性が探索された。FIRMは、再生医療分野の唯一の業界団体として、まだ萌芽的な段階にある再生医療分野における参加企業間の共通言語の構築や問題意識の調整、業界としての要望の取りまとめと意見表明などの対応を通じて、文字通り「再生医療の研究成果を、社会に対して、安全かつ安定的に提供できる体制を構築…(中略)…再生医療専業化の道筋を示す」[10]ためのさまざまな働きかけを行ってきた。
最終的には、日本再生医療学会とFIRMの動き、そして行政側のニーズとアクションがうまく調和することで、早期の実用化と安全性確保のための法制化が進むこととなった。先述のハートシートはこうした制度改革の結果として承認に至った事例ということができよう。こうしたアカデミアや業界団体の動きに呼応する形で、2012年7月に経済産業省が「再生医療の実用化・産業化に関する研究会」(事務局:経済産業省製造産業局生物化学産業課)を設置することにより、実用化・産業化に向けた産官学それぞれの問題意識や要望を具体的な政策課題へと落とし込み、そしてコンセンサスを得る場が構築された。2013年2月にとりまとめられた報告書(最終取りまとめ)では、「必要な制度的枠組み」として①医療機関から外部の事業者に委託できる制度の整備の必要性、②医薬品の審査・承認手続きとは異なる再生医療の特性を踏まえた安全性基準の整備と早期承認制度の導入の2点が提示されているが、これらの論点はほぼそのまま再生医療安全確保法および薬事法改正に反映されることになった[11]。行政による個別的なヒアリングを通じてアカデミアや業界団体といったステークホルダーの意向や要望を収集したり、その反対にステークホルダーの側がある種のロビイング(ロビー活動)を行うことで非公式に意向や要望を伝達したりするといったプロセスは従来からみられてきたが、「再生医療の実用化・産業化に関する研究会」の例にみられるように、公式の場において「規制のラグ」の克服に向けて産学の立場から様々な意見が提示され、その調整が行われることで新たな規制のフレームワークが検討されるといったスキームは当時としては非常にユニークな試みであったといえる[12]。
5. 協調的な規制に潜むリスク
再生医療法制の整備過程は、規制主体と被規制主体との協調的な規制の形成による「規制のラグ」の克服を目指した事例といえる。では、「規制のラグ」の克服手段として、こうした被規制主体と規制主体の協調的な規制形成が万能なのかといえば、必ずしもそうではない。協調的な規制政策の形成には、かねてより「規制の虜」(Regulatory Capture)のリスクが指摘されてきた[13]。
たとえばエネルギー政策の分野において、電力事業者を規制するはずの政府が、事業者側の情報優位を背景にむしろ電力会社によって取りこまれてしまっている状況が指摘されたように[14]、規制主体と被規制主体との協調的な関係性が「一種の腐敗というべきキャプチャー」(corrosive capture)ともいえる関係性に転じうることは想像に難くない[15]。規制によって「仕切られた市場」である医療分野は、こうした政府とアカデミア・企業(あるいは業界団体)との関係性が近いがゆえに、こうした「規制の虜」のリスクが少なからず存在するはずである。したがって、こうした官民の協調的な関係性が「腐敗」に転じることなく、一定の緊張関係を保つことで新たな規制手段を講じるために必要な情報提供やそのためのコミュニケーションとして機能するよう「健全な」関係性を規律することが求められるのである。
なお、2014年の再生医療法制の整備、特に世界的にも例のない早期実用化のための仕組みであった「条件・期限付承認」の導入が、こうしたcorrosive captureにあたるのかという点については別途慎重な検証が求められよう。しかしながら、2024年2月現在においてこうした「条件・期限付き承認」が何らかの重大な事故や事件を誘発したという報告がみられていないことを考えると、早期実用化のための制度設計がアカデミアや企業といった研究開発サイドのインセンティブやモチベーションを優先した結果として患者のリスクを増大させた、とはいえないのではないかと考えられる。
6. 「規制の虜」を克服する手段としての「レスポンシブル・ロビイング」
既に述べたように、医療分野は伝統的な規制政策の領域でありながら、官民関係の変化とそれに伴う協調的な規制の形成という試みが既に観察されつつある。重要なのは、どのようにすれば「腐敗」に陥ることなく協調的な関係性が機能するかである。そのヒントとなるのが「レスポンシブル・ロビイング」(responsible lobbying)という概念である。規制の立案プロセスに対して企業やアカデミアがその社会的責務として関与するという考え方であり、被規制主体の側には個別的な利益の最大化や短期的な利益誘導というモチベーションを超えた公共政策への貢献姿勢・能力が求められる。それは、いわば民間企業あるいは業界団体を主体とした科学的助言の一形態というべきものである[16]。
現在、必ずしも多くの組織資源が割かれていないと思われる企業あるいは業界団体のガバメント・アフェアーズやパブリック・アフェアーズは、まさにこうした役割を担うことが期待される組織といえる。IT等の新興テクノロジー分野において、近年、企業内における公共政策部門の組織資源と活動量が大きくなっているのに比べて、医療をはじめとする伝統的な技術分野においては必ずしもこうした機能強化が進んでいないように見受けられる。これらの組織による責任ある関与と情報提供こそが的確でタイムリーな規制形成の成否を分かつと考えられる。そのため、自社および業界団体の現在、そして将来をとりまく政策環境に対する積極的な関与がこれまで以上に求められている。無論、こうした被規制主体の側による政策過程における責任ある関与は、決してボランタリーな行動たりえない。新興科学技術に対する新たな規制の形成過程への企業あるいは業界団体による関与と貢献を促す上では、行政側による的確なインセンティブの設計が不可欠である。
一方で、被規制主体によるこうした政策過程への積極的な関与を巡っては、やはり「キャプチャー」の可能性についても留意しなければならない。被規制主体の情報優位は、規制主体の行動に対する一定の制御可能性をもたらす。「責任あるロビイング」が「規制の虜」に陥ることなく、規制主体と被規制主体が健全な緊張関係と協調関係のなかで新たな規制の導入や既存の規制の変更に向けた検討を行っていくためには、予防方策として新たな官民関係のあり方の整理とそれぞれの行動規範の明確化が求められる。先述の「再生医療の実用化・産業化に関する研究会」の事例や、2021年(令和3)度に日本医療研究開発機構(AMED)において推進された「医療機器開発ガイドラインの今後のあり方に関する調査」[17]および2022年(令和4)度の「医療機器開発ガイドライン(手引き)のためのガイドライン策定調査」[18]など、近年取り組まれている新たなガイドライン策定に向けた官民間のコミュニケーションは一定の透明性を持った形で進められつつある。こうしたコミュニケーションの透明化というアプローチは「規制の虜」のリスクを軽減する有効な手法の一つであると考えられる。また、特定の事業者や団体による情報提供に依存するのではなく、より多元的な主体が規制の形成過程に関与し、いわば情報提供の質を競い合うことで、結果としてはより良い規制の実現につながるものと考えられる[19]。こうした透明性と競争性のある形での官民間のコミュニケーションの制度化は、「レスポンシブル・ロビイング」を実現するための有用な方策の一つとして期待される。
<参考文献>
[1] 本稿は、黒河昭雄「第4章 科学技術政策における官民関係の変容-医療を事例として」『政策研究:社会課題対応のための科学技術政策システムの再構築』東京財団政策研究所(2024年3月)の内容をもとに一部を加筆・再構成したものである。
[2] 黒河昭雄(2022)「革新的な製品・サービスに対する新たな規制の導入プロセスに関するケーススタディ ―AI医療機器を事例に(上)(下)」『Review』, 2022年2月,
https://www.tkfd.or.jp/research/detail.php?id=3928, https://www.tkfd.or.jp/research/detail.php?id=3929
[3] 黒河昭雄(2023)「レギュラトリーリスクと制度的な公正性 -がん免疫治療薬「オプジーボ」をめぐる緊急薬価改定を事例に」『Review』, 2023年6月, https://www.tkfd.or.jp/research/detail.php?id=4270
[4] S. Kano, "Interaction analysis between innovation and regulation: The concept of Regulatory Science as a Process (RaaP) and its applications," 2016 Portland International Conference on Management of Engineering and Technology (PICMET), Honolulu, HI, USA, 2016, pp. 1278-1285, doi: 10.1109/PICMET.2016.7806704.
[5] 大阪大学大学院医学系研究科・医学部WEBサイト「NEWS & TOPICS 大阪大学とテルモ株式会社が再生医療等製品「ハートシート」の共同開発で「産学官連携功労者表彰 厚生労働大臣賞」を受賞しました」https://www.med.osaka-u.ac.jp/archives/2527(2024年2月5日アクセス)
[6] テルモWEBサイト「テルモ、『ハートシート』の製造販売承認を取得」(2015年9月18日)https://www.terumo.co.jp/newsrelease/detail/20150918/732(2024年2月5日アクセス)
[7] 日本再生医療学会が2013年5月8日に出した提言・声明「再生医療推進法国会成立報告とさらなる推進に向けて」では、「2013年4月26日に議員立法の再生医療推進法が成立いたしました。この法制化は、これまでの日本再生医療学会による政治や行政との連携協力、および再生医療実現に向けた積極的な諸活動によるものです。今後の再生医療安全性確保等法と改正薬事法の成立によって、本格的な再生医療の推進が可能となります」とされており、学会による積極的な政策形成への関与が示唆されている。
[8] 厚生労働省(2012)「厚生科学審議会科学技術部会再生医療の安全性確保と推進に関する専門委員会 第2回資料」2012年11月16日、https://www.mhlw.go.jp/stf/shingi/2r9852000002l0m5.html(2024年2月5日アクセス)
[9] 日本再生医療学会(2011)「会員の皆様へのお願い(勧告)」2011年3月1日, https://www.jsrm.jp/news/news-649/」
[10] 一般社団法人再生医療イノベーションフォーラムWEBサイト「自主基準」https://firm.or.jp/about/voluntary-standards/(2024年2月5日アクセス)
[11] 経済産業省(2013)「再生医療の実用化・産業化に関する報告書 最終とりまとめ」2013年2月, https://www.mhlw.go.jp/stf/shingi/2r9852000002v591-att/2r9852000002v5dn.pdf
[12] もっとも、こうした透明性を持った公式の「場」が作られるということは、同時にさらに非公式な「場」が形成されることをも意味すると考えられる。
[13] Carpenter, D., & Moss, D. (Eds.). (2013). Preventing Regulatory Capture: Special Interest Influence and How to Limit it. Cambridge: Cambridge University Press. doi:10.1017/CBO9781139565875;
[14] 黒川清(2016)『規制の虜 グループシンクが日本を滅ぼす』講談社, 2016年3月.
[15] 村上裕一(2016)「いわゆる Corrosive Capture とその予防方策」年報 公共政策学, 10, 141-16, 2016年3月.
[16] 岸本 充生(2024)「責任ある研究・イノベーション(RRI)としての『レスポンシブル・ロビイング』」『Review』, 2024年2月, https://www.tkfd.or.jp/research/detail.php?id=4422
[17] 株式会社ドゥリサーチ研究所(2022)「医療機器開発ガイドラインの今後のあり方に関する調査 調査報告書」2022年3月, https://www.amed.go.jp/content/000101886.pdf(2024年3月27日アクセス)
[18] 株式会社ドゥリサーチ研究所(2023)「医療機器開発ガイドライン(手引き)のためのガイドライン策定調査報告書」2023年3月, https://www.amed.go.jp/content/000115176.pdf(2024年3月27日アクセス)
[19] なお、規制の執行部門であるPMDAにおいても、2012年に設置された科学委員会を通じて外部(アカデミア)の知見の獲得が試みられている。AI医療機器についてはこの科学委員会におかれた専門部会が実質的なルール形成のアジェンダ設定の役割を担った。詳しくは、黒河(2022, 前掲)を参照されたい。
-
-

- 元 主任研究員
- 黒河 昭雄
- 黒河 昭雄
- 研究分野・主な関心領域
-
- 公共政策
- 医療イノベーション政策
- 科学技術イノベーション政策
- 政策のための科学
-
-
- 神奈川県立保健福祉大学ヘルスイノベーション研究科 教授
- 昌子 久仁子
- 昌子 久仁子

注目コンテンツ
-
2025年の年金改正のポイント
2025年の年金改正のポイント
-
2025年の日本で気にすべきは、インフレか、それともデフレか -政府はデフレを気にし、日本銀行はインフレを気にしている?-
2025年の日本で気にすべきは、インフレか、それともデフレか -政府はデフレを気にし、日本銀行はインフレを気にしている?-
-
れいわ新選組「消費税ゼロ」の実現可能性を探る- 連載コラム「税の交差点」第71回
れいわ新選組「消費税ゼロ」の実現可能性を探る- 連載コラム「税の交差点」第71回
-
r-g(金利と成長率の差)はどこまで上昇し得るのか? -既存研究のReviewとシミュレーションからの示唆-
r-g(金利と成長率の差)はどこまで上昇し得るのか? -既存研究のReviewとシミュレーションからの示唆-
-
《時評》人体の不思議展と先端手術研修~人の尊厳と遺体の扱いについて~(2009年7月17日再改訂)
《時評》人体の不思議展と先端手術研修~人の尊厳と遺体の扱いについて~(2009年7月17日再改訂)








































.jpg)
